7 Differential Expression
There are many different methods for calculating differential expression between groups in scRNAseq data. There are a number of review papers worth consulting on this topic.
There is the Seurat differential expression Vignette which walks through the variety implemented in Seurat.
There is also a good discussion of useing pseudobulk approaches which is worth checking out if youre planning differential expression analyses.
We will now look at GSE96583, another PBMC dataset. For speed, we will be looking at a subset of 5000 cells from this data. The cells in this dataset were pooled from eight individual donors. This data contains two batches of single cell sequencing. One of the batches was stimulated with IFN-beta.
The data has already been processed as we have done with the first PBMC dataset, and can be loaded from the kang2018.rds file in the data folder.
kang <- readRDS("data/kang2018.rds")
head(kang@meta.data)
#> orig.ident nCount_RNA nFeature_RNA ind
#> AGGGCGCTATTTCC-1 SeuratProject 2020 523 1256
#> GGAGACGATTCGTT-1 SeuratProject 840 381 1256
#> CACCGTTGTCGTAG-1 SeuratProject 3097 995 1016
#> TATCGTACACGCAT-1 SeuratProject 1011 540 1488
#> TGACGCCTTGCTTT-1 SeuratProject 570 367 101
#> TACGAGACCTATTC-1 SeuratProject 2399 770 1244
#> stim cell multiplets
#> AGGGCGCTATTTCC-1 stim CD14+ Monocytes singlet
#> GGAGACGATTCGTT-1 stim CD4 T cells singlet
#> CACCGTTGTCGTAG-1 ctrl FCGR3A+ Monocytes singlet
#> TATCGTACACGCAT-1 stim B cells singlet
#> TGACGCCTTGCTTT-1 ctrl CD4 T cells ambs
#> TACGAGACCTATTC-1 stim CD4 T cells singletHow cells from each condition do we have?
table(kang$stim)
#>
#> ctrl stim
#> 2449 2551How many cells per individuals per group?
table(kang$ind, kang$stim)
#>
#> ctrl stim
#> 101 178 229
#> 107 117 107
#> 1015 514 496
#> 1016 356 356
#> 1039 100 118
#> 1244 380 313
#> 1256 394 390
#> 1488 410 542And for each sample, how many of each cell type has been classified?
table(paste(kang$ind,kang$stim), kang$cell)
#>
#> B cells CD14+ Monocytes CD4 T cells CD8 T cells
#> 101 ctrl 24 48 61 15
#> 101 stim 30 52 84 17
#> 1015 ctrl 81 149 145 46
#> 1015 stim 68 151 150 22
#> 1016 ctrl 22 72 89 112
#> 1016 stim 29 65 66 115
#> 1039 ctrl 7 35 40 6
#> 1039 stim 7 28 51 6
#> 107 ctrl 9 51 32 6
#> 107 stim 9 35 43 1
#> 1244 ctrl 23 86 206 8
#> 1244 stim 18 58 191 4
#> 1256 ctrl 32 81 180 29
#> 1256 stim 42 70 198 25
#> 1488 ctrl 36 59 247 13
#> 1488 stim 59 59 325 15
#>
#> Dendritic cells FCGR3A+ Monocytes
#> 101 ctrl 4 11
#> 101 stim 6 23
#> 1015 ctrl 4 50
#> 1015 stim 17 44
#> 1016 ctrl 4 22
#> 1016 stim 2 32
#> 1039 ctrl 1 3
#> 1039 stim 1 8
#> 107 ctrl 3 12
#> 107 stim 2 5
#> 1244 ctrl 8 19
#> 1244 stim 6 4
#> 1256 ctrl 6 20
#> 1256 stim 3 11
#> 1488 ctrl 8 25
#> 1488 stim 12 28
#>
#> Megakaryocytes NK cells
#> 101 ctrl 4 11
#> 101 stim 1 16
#> 1015 ctrl 5 34
#> 1015 stim 5 39
#> 1016 ctrl 4 31
#> 1016 stim 1 46
#> 1039 ctrl 6 1
#> 1039 stim 10 5
#> 107 ctrl 1 3
#> 107 stim 0 12
#> 1244 ctrl 5 25
#> 1244 stim 4 28
#> 1256 ctrl 1 45
#> 1256 stim 8 33
#> 1488 ctrl 4 18
#> 1488 stim 6 387.1 Prefiltering
Why do we need to do this?
If expression is below a certain level, it will be almost impossible to see any differential expression.
When doing differential expression, you generally ignore genes with low expression. In single cell datasets, there are many genes like this. Filtering here to make our dataset smaller so it runs quicker, and there is less aggressive correction for multiple hypotheses.
How many genes before filtering?
kang
#> An object of class Seurat
#> 35635 features across 5000 samples within 1 assay
#> Active assay: RNA (35635 features, 2000 variable features)
#> 3 layers present: counts, data, scale.data
#> 2 dimensional reductions calculated: pca, umapHow many copies of each gene are there?
total_per_gene <- rowSums(GetAssayData(kang, 'RNA', layer = "counts")) #Make sure its RNA assay, layer = counts
hist(log10(total_per_gene))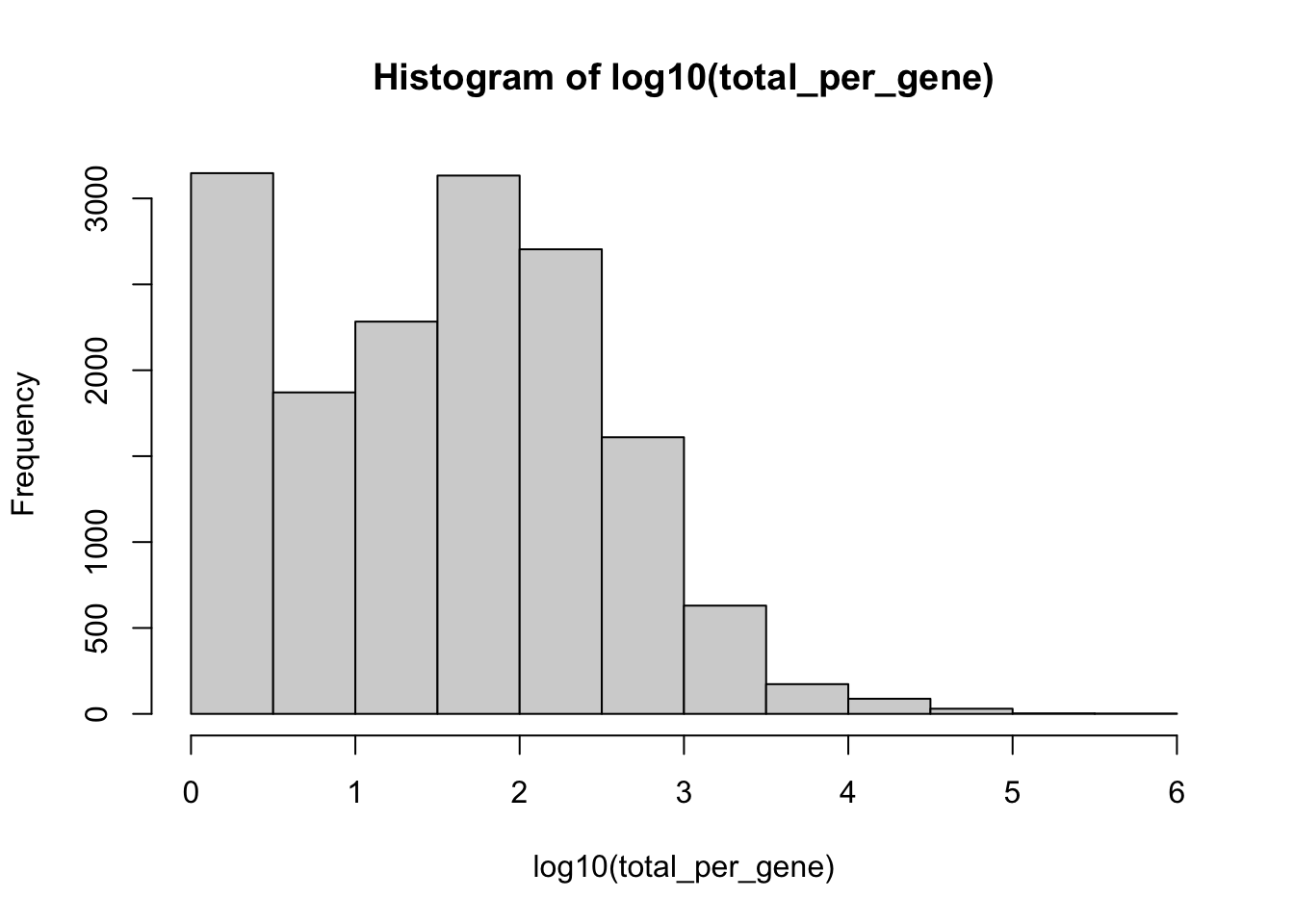
Lets keep only those genes with at least 50 copies across the entire experiment.
kang <- kang[total_per_gene >= 50, ] How many genes after filtering?
kang
#> An object of class Seurat
#> 7216 features across 5000 samples within 1 assay
#> Active assay: RNA (7216 features, 1228 variable features)
#> 3 layers present: counts, data, scale.data
#> 2 dimensional reductions calculated: pca, umapWe might like to see the effect of IFN-beta stimulation on each cell type individually. For the purposes of this workshop, just going to test one cell type; CD14+ Monocytes
An easy way is to subset the object.
kang.celltype <- kang[, kang$cell == "CD14+ Monocytes" ]
DimPlot(kang.celltype)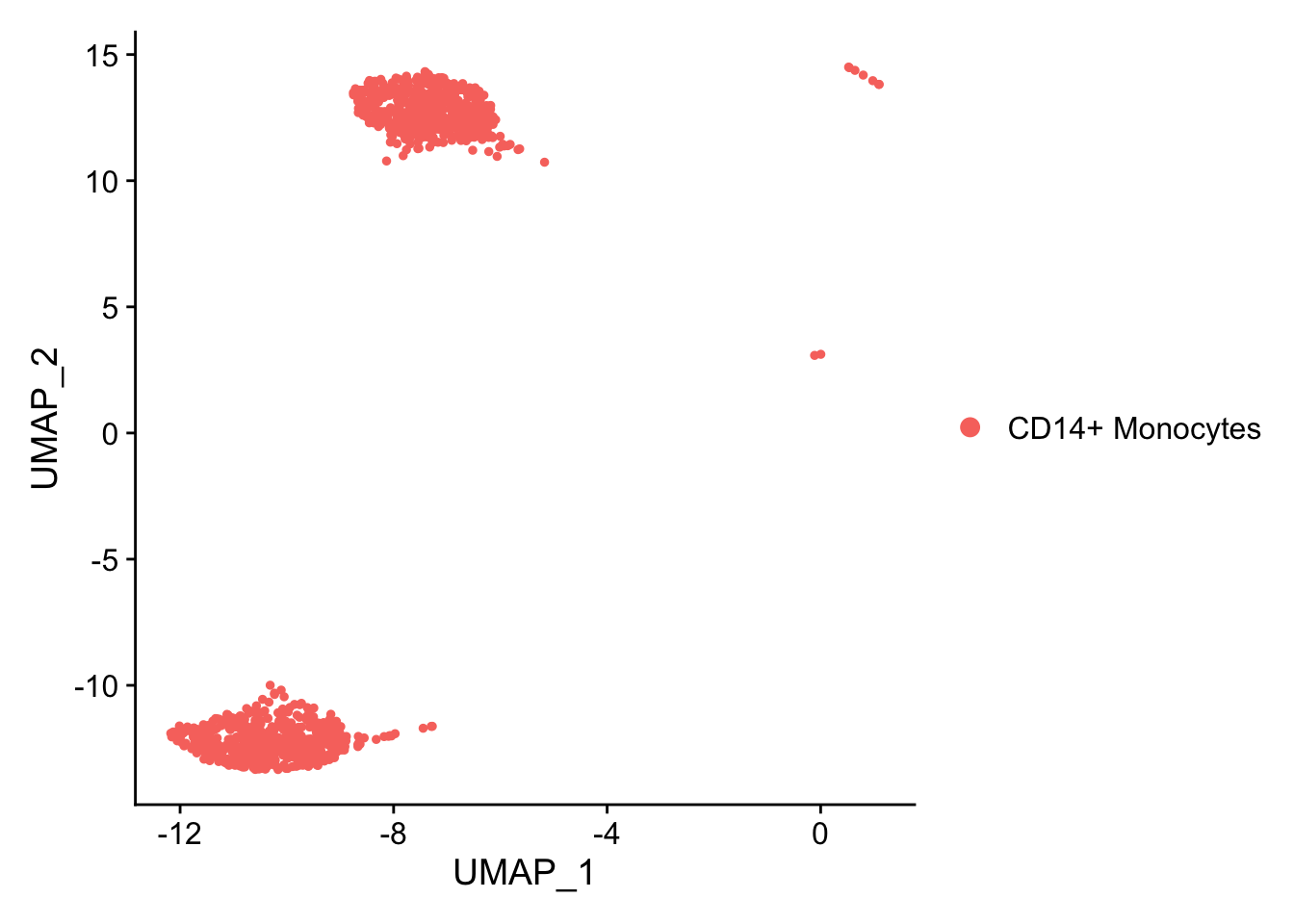
7.2 Default Wilcox test
To run this test, we change the Idents to the factor(column) we want to test. In this case, that’s ‘stim’.
# Change Ident to Condition
Idents(kang.celltype) <- kang.celltype$stim
# default, wilcox test
de_result_wilcox <- FindMarkers(kang.celltype,
ident.1 = 'stim',
ident.2 = 'ctrl',
logfc.threshold = 0, # Give me ALL results
min.pct = 0
)
#> For a (much!) faster implementation of the Wilcoxon Rank Sum Test,
#> (default method for FindMarkers) please install the presto package
#> --------------------------------------------
#> install.packages('devtools')
#> devtools::install_github('immunogenomics/presto')
#> --------------------------------------------
#> After installation of presto, Seurat will automatically use the more
#> efficient implementation (no further action necessary).
#> This message will be shown once per session
# Add average expression for plotting
de_result_wilcox$AveExpr<- rowMeans(GetAssayData(kang.celltype,assay="RNA", layer = "data")[rownames(de_result_wilcox),])Look at the top differentially expressed genes.
head(de_result_wilcox)
#> p_val avg_log2FC pct.1 pct.2 p_val_adj
#> RSAD2 5.541857e-197 6.807428 0.975 0.043 3.999004e-193
#> CXCL10 9.648067e-197 8.043584 0.973 0.038 6.962045e-193
#> IFIT3 4.988121e-196 6.232966 0.979 0.050 3.599428e-192
#> TNFSF10 1.116418e-195 6.567507 0.977 0.055 8.056075e-192
#> IFIT1 8.116699e-190 6.764859 0.950 0.026 5.857010e-186
#> ISG15 1.524836e-187 6.373685 0.998 0.320 1.100322e-183
#> AveExpr
#> RSAD2 1.686530
#> CXCL10 2.388462
#> IFIT3 1.701247
#> TNFSF10 1.682688
#> IFIT1 1.584751
#> ISG15 3.239774
p1 <- ggplot(de_result_wilcox, aes(x=AveExpr, y=avg_log2FC, col=p_val_adj < 0.05)) +
geom_point() +
scale_colour_manual(values=c('TRUE'="red",'FALSE'="black")) +
theme_bw() +
ggtitle("Wilcox Test")
p2 <- ggplot(de_result_wilcox, aes(x=avg_log2FC, y=-log10(p_val), col=p_val_adj < 0.05)) +
geom_point() +
scale_colour_manual(values=c('TRUE'="red",'FALSE'="black")) +
theme_bw() +
ggtitle("Wilcox Test (Volcano)")
p1 + p2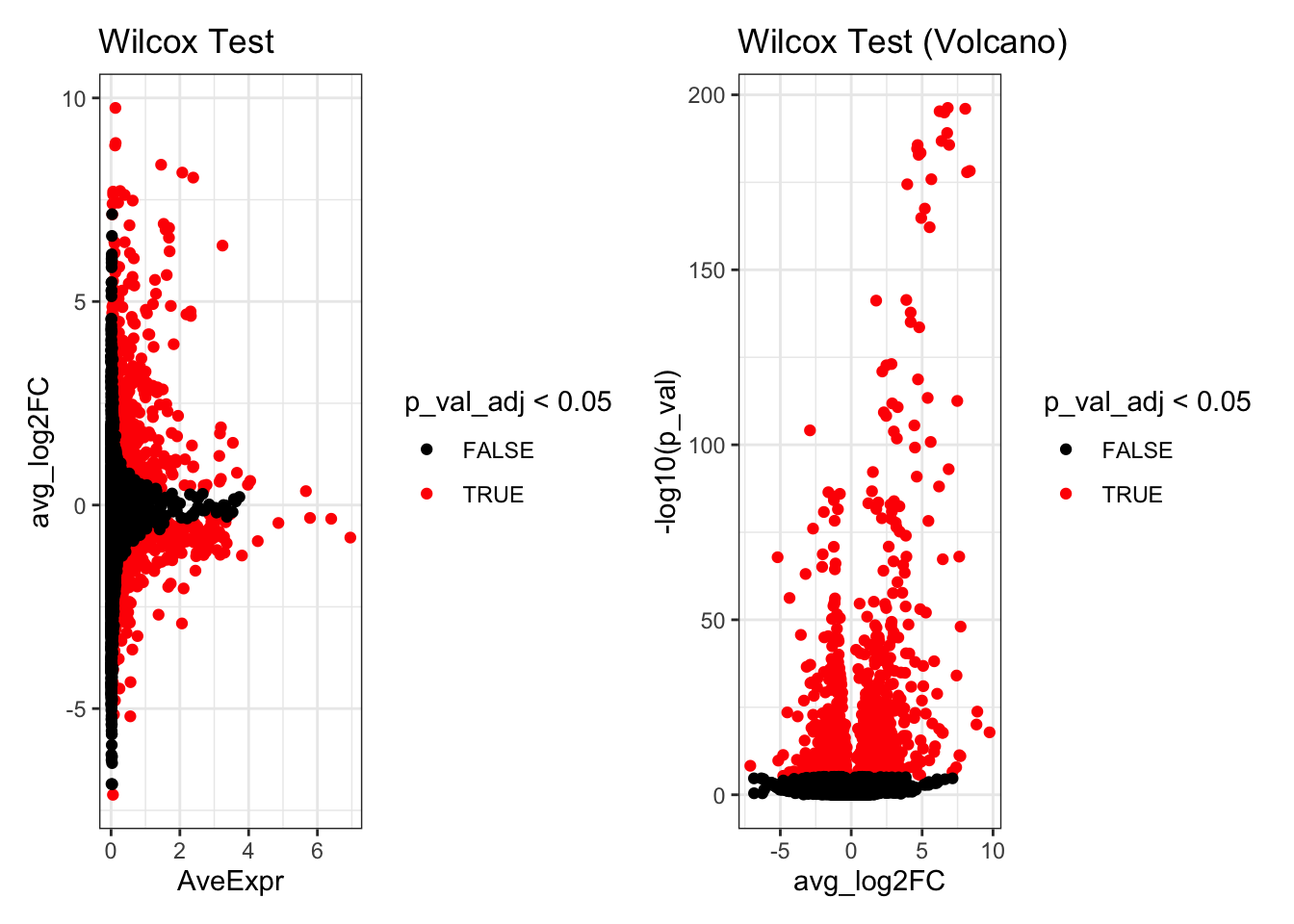
7.3 Seurat Negative binomial
Negative binonial test is run almost the same way - just need to specify it under ‘test.use’
# Change Ident to Condition
Idents(kang.celltype) <- kang.celltype$stim
# default, wilcox test
de_result_negbinom <- FindMarkers(kang.celltype,
test.use="negbinom", # Choose a different test.
ident.1 = 'stim',
ident.2 = 'ctrl',
logfc.threshold = 0, # Give me ALL results
min.pct = 0
)
# Add average expression for plotting
de_result_negbinom$AveExpr<- rowMeans(GetAssayData(kang.celltype,assay="RNA", layer = "data")[rownames(de_result_negbinom),])Look at the top differentially expressed genes.
head(de_result_negbinom)
#> p_val avg_log2FC pct.1 pct.2 p_val_adj AveExpr
#> CXCL10 0 8.043584 0.973 0.038 0 2.388462
#> CCL8 0 8.166731 0.909 0.022 0 2.071022
#> LY6E 0 4.891032 0.973 0.133 0 1.736931
#> APOBEC3A 0 4.755297 0.992 0.258 0 2.311961
#> IFITM3 0 4.683573 0.994 0.267 0 2.190185
#> IFI6 0 3.950817 0.979 0.253 0 1.820140
p1 <- ggplot(de_result_negbinom, aes(x=AveExpr, y=avg_log2FC, col=p_val_adj < 0.05)) +
geom_point() +
scale_colour_manual(values=c('TRUE'="red",'FALSE'="black")) +
theme_bw() +
ggtitle("Negative Bionomial Test")
p2 <- ggplot(de_result_negbinom, aes(x=avg_log2FC, y=-log10(p_val), col=p_val_adj < 0.05)) +
geom_point() +
scale_colour_manual(values=c('TRUE'="red",'FALSE'="black")) +
theme_bw() +
ggtitle("Negative Bionomial Test (Volcano)")
p1 + p2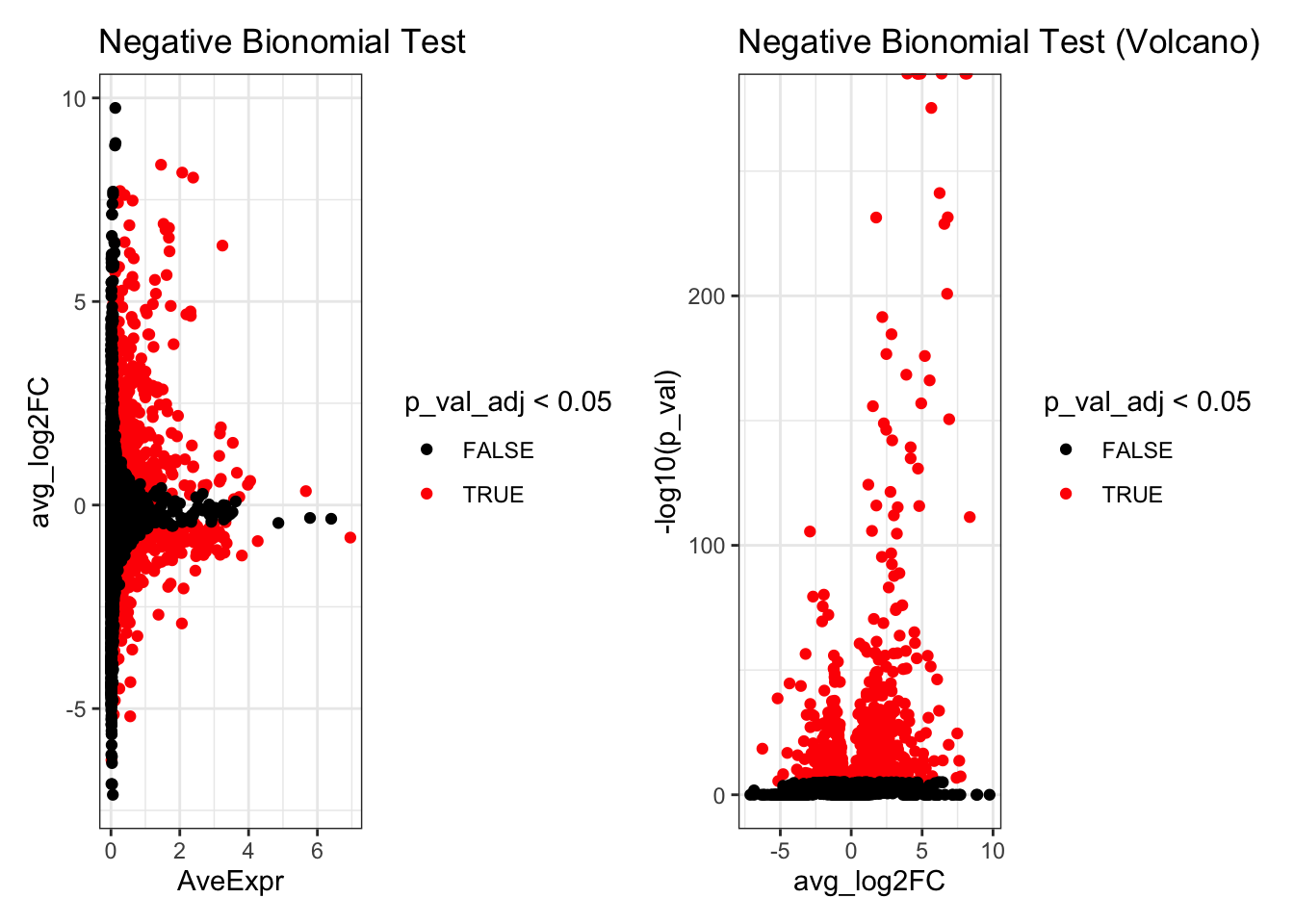
7.4 Pseudobulk
Pseudobulk analysis is an option where you have biological replicates. It is essentially pooling the individual cell counts and treating your expreiment like a bulk RNAseq.
First, you need to build a pseudobulk matrix - the AggregateExpression() function can do this, once you set the ‘Idents’ of your seurat object to your grouping factor (here, thats a combination of individual+treatment called ‘sample’, instead of the ‘stim’ treatment column).
# Tools for bulk differential expression
library(limma)
#>
#> Attaching package: 'limma'
#> The following object is masked from 'package:BiocGenerics':
#>
#> plotMA
library(edgeR)
#>
#> Attaching package: 'edgeR'
#> The following object is masked from 'package:SingleCellExperiment':
#>
#> cpm
# Change idents to ind for grouping.
kang.celltype$sample <- factor(paste(kang.celltype$stim, kang.celltype$ind, sep="_"))
Idents(kang.celltype) <- kang.celltype$sample
# THen pool together counts in those groups
# AggregateExperssion returns a list of matricies - one for each assay requested (even just requesting one)
pseudobulk_matrix_list <- AggregateExpression( kang.celltype, assays='RNA')
#> Names of identity class contain underscores ('_'), replacing with dashes ('-')
#> This message is displayed once every 8 hours.
pseudobulk_matrix <- pseudobulk_matrix_list[['RNA']]
colnames(pseudobulk_matrix) <- as.character(colnames(pseudobulk_matrix)) # Changes colnames to simple text
pseudobulk_matrix[1:5,1:4]
#> 5 x 4 sparse Matrix of class "dgCMatrix"
#> ctrl-101 ctrl-1015 ctrl-1016 ctrl-1039
#> NOC2L 2 7 . .
#> HES4 . 3 2 1
#> ISG15 31 185 236 41
#> TNFRSF18 . 3 4 2
#> TNFRSF4 . 2 . .
# Aggregate expression replaces _ with -! We're going to change it back (for limma.)
colnames(pseudobulk_matrix) <- gsub("-", "_",colnames(pseudobulk_matrix))Now it looks like a bulk RNAseq experiment, so treat it like one.
We can use the popular limma package for differential expression. Here is one tutorial, and the hefty reference manual is hosted by bioconductor.
In brief, this code below constructs a linear model for this experiment that accounts for the variation in individuals and treatment. It then tests for differential expression between ‘stim’ and ‘ctrl’ groups.
dge <- DGEList(pseudobulk_matrix)
dge <- calcNormFactors(dge)
# Remove _ and everything after it - yeilds stim group
stim <- gsub("_.*","",colnames(pseudobulk_matrix))
# Removing everything before the _ for the individua, then converting those numerical ind explictiy to text. Else limma will treat them as numbers!
ind <- as.character(gsub(".*_","",colnames(pseudobulk_matrix)))
design <- model.matrix( ~0 + stim + ind)
vm <- voom(dge, design = design, plot = FALSE)
fit <- lmFit(vm, design = design)
contrasts <- makeContrasts(stimstim - stimctrl, levels=coef(fit))
fit <- contrasts.fit(fit, contrasts)
fit <- eBayes(fit)
de_result_pseudobulk <- topTable(fit, n = Inf, adjust.method = "BH")
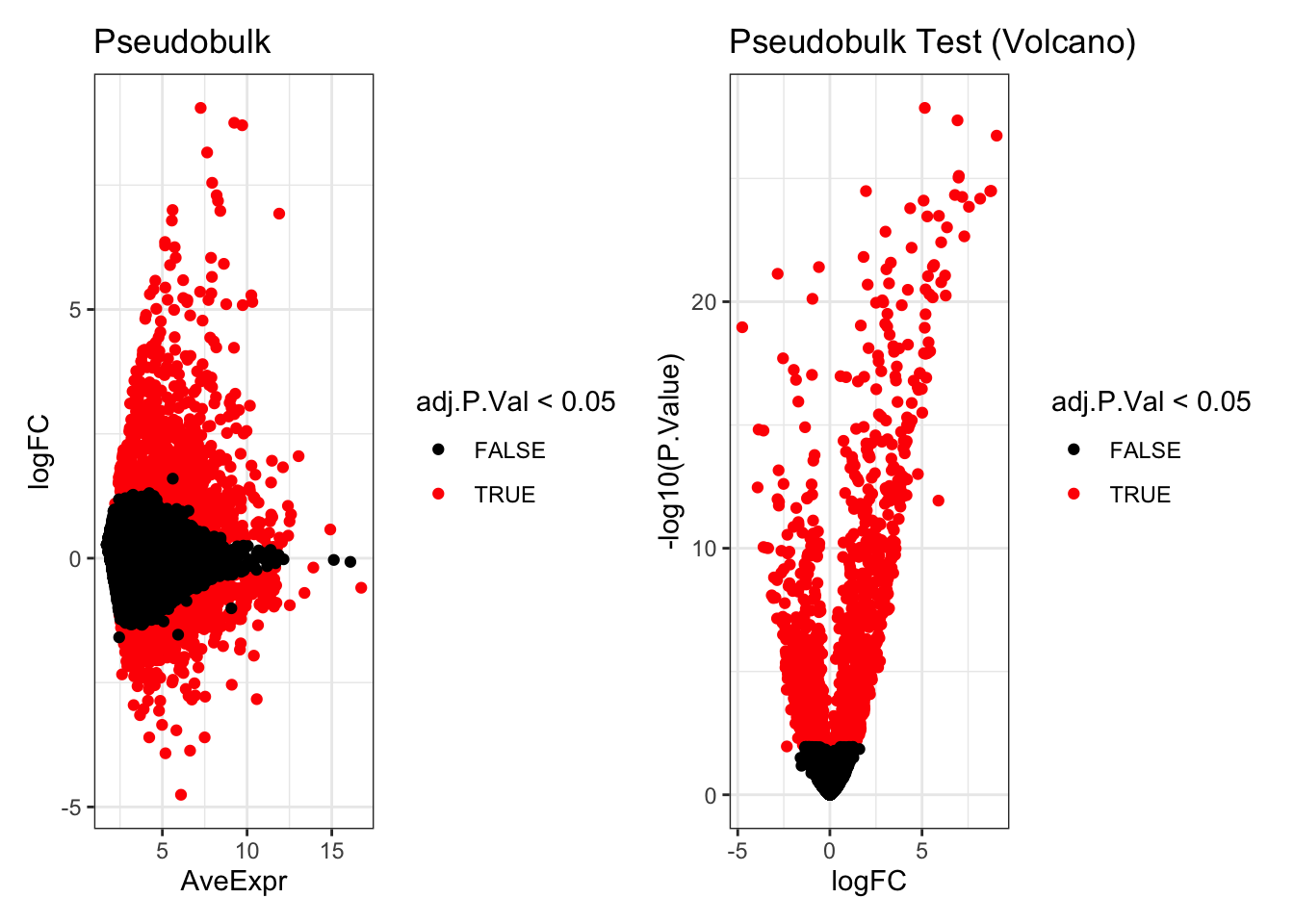
de_result_pseudobulk <- arrange(de_result_pseudobulk , adj.P.Val)Look at the significantly differentially expressed genes:
head(de_result_pseudobulk)
#> logFC AveExpr t P.Value
#> ISG20 5.151090 10.311187 34.62460 1.377577e-28
#> ISG15 6.926462 11.895928 33.45672 4.402969e-28
#> CXCL11 9.051653 7.260525 32.07090 1.838679e-27
#> IFIT3 6.980913 8.420719 28.54319 9.234346e-26
#> HERC5 6.998957 5.602349 28.68162 7.853707e-26
#> TMSB10 1.959063 11.466981 27.48469 3.264041e-25
#> adj.P.Val B
#> ISG20 9.940598e-25 55.27733
#> ISG15 1.588591e-24 54.12183
#> CXCL11 4.422636e-24 51.56638
#> IFIT3 1.332701e-22 48.32088
#> HERC5 1.332701e-22 48.02111
#> TMSB10 2.950103e-22 47.34483
p1 <- ggplot(de_result_pseudobulk, aes(x=AveExpr, y=logFC, col=adj.P.Val < 0.05)) +
geom_point() +
scale_colour_manual(values=c('TRUE'="red",'FALSE'="black")) +
theme_bw() +
ggtitle("Pseudobulk")
p2 <- ggplot(de_result_pseudobulk, aes(x=logFC, y=-log10(P.Value), col=adj.P.Val < 0.05)) +
geom_point() +
scale_colour_manual(values=c('TRUE'="red",'FALSE'="black")) +
theme_bw() +
ggtitle("Pseudobulk Test (Volcano)")
p1 + p2