6 SingleR
#install.packages("BiocManager")
#BiocManager::install(c("SingleCellExperiment","SingleR","celldex"),ask=F)
library(SingleCellExperiment)
library(SingleR)
library(celldex)In this workshop we have focused on the Seurat package. However, there is another whole ecosystem of R packages for single cell analysis within Bioconductor. We won’t go into any detail on these packages in this workshop, but there is good material describing the object type online : OSCA.
For now, we’ll just convert our Seurat object into an object called SingleCellExperiment. Some popular packages from Bioconductor that work with this type are Slingshot, Scran, Scater.
sce <- as.SingleCellExperiment(pbmc)
sce
#> class: SingleCellExperiment
#> dim: 13714 2638
#> metadata(0):
#> assays(3): counts logcounts scaledata
#> rownames(13714): AL627309.1 AP006222.2 ... PNRC2.1
#> SRSF10.1
#> rowData names(0):
#> colnames(2638): AAACATACAACCAC-1 AAACATTGAGCTAC-1 ...
#> TTTGCATGAGAGGC-1 TTTGCATGCCTCAC-1
#> colData names(31): orig.ident nCount_RNA ...
#> pretty_cluster_labels ident
#> reducedDimNames(2): PCA UMAP
#> mainExpName: RNA
#> altExpNames(0):We will now use a package called SingleR to label each cell. SingleR uses a reference data set of cell types with expression data to infer the best label for each cell. A convenient collection of cell type reference is in the celldex package which currently contains the follow sets:
ls('package:celldex')
#> [1] "BlueprintEncodeData"
#> [2] "DatabaseImmuneCellExpressionData"
#> [3] "HumanPrimaryCellAtlasData"
#> [4] "ImmGenData"
#> [5] "MonacoImmuneData"
#> [6] "MouseRNAseqData"
#> [7] "NovershternHematopoieticData"In this example, we’ll use the HumanPrimaryCellAtlasData set, which contains high-level, and fine-grained label types. Lets download the reference dataset
# This too is a sce object,
# colData is equivalent to seurat's metadata
ref.set <- celldex::HumanPrimaryCellAtlasData()
#> snapshotDate(): 2023-04-24
#> see ?celldex and browseVignettes('celldex') for documentation
#> loading from cache
#> see ?celldex and browseVignettes('celldex') for documentation
#> loading from cacheThe “main” labels.
unique(ref.set$label.main)
#> [1] "DC" "Smooth_muscle_cells"
#> [3] "Epithelial_cells" "B_cell"
#> [5] "Neutrophils" "T_cells"
#> [7] "Monocyte" "Erythroblast"
#> [9] "BM & Prog." "Endothelial_cells"
#> [11] "Gametocytes" "Neurons"
#> [13] "Keratinocytes" "HSC_-G-CSF"
#> [15] "Macrophage" "NK_cell"
#> [17] "Embryonic_stem_cells" "Tissue_stem_cells"
#> [19] "Chondrocytes" "Osteoblasts"
#> [21] "BM" "Platelets"
#> [23] "Fibroblasts" "iPS_cells"
#> [25] "Hepatocytes" "MSC"
#> [27] "Neuroepithelial_cell" "Astrocyte"
#> [29] "HSC_CD34+" "CMP"
#> [31] "GMP" "MEP"
#> [33] "Myelocyte" "Pre-B_cell_CD34-"
#> [35] "Pro-B_cell_CD34+" "Pro-Myelocyte"An example of the types of “fine” labels.
head(unique(ref.set$label.fine))
#> [1] "DC:monocyte-derived:immature"
#> [2] "DC:monocyte-derived:Galectin-1"
#> [3] "DC:monocyte-derived:LPS"
#> [4] "DC:monocyte-derived"
#> [5] "Smooth_muscle_cells:bronchial:vit_D"
#> [6] "Smooth_muscle_cells:bronchial"Now we’ll label our cells using the SingleCellExperiment object, with the above reference set.
pred.cnts <- SingleR::SingleR(test = sce, ref = ref.set, labels = ref.set$label.main)Keep any types that have more than 10 cells to the label, and put those labels back on our Seurat object and plot our on our umap.
lbls.keep <- table(pred.cnts$labels)>10
pbmc$SingleR.labels <- ifelse(lbls.keep[pred.cnts$labels], pred.cnts$labels, 'Other')
DimPlot(pbmc, reduction='umap', group.by='SingleR.labels')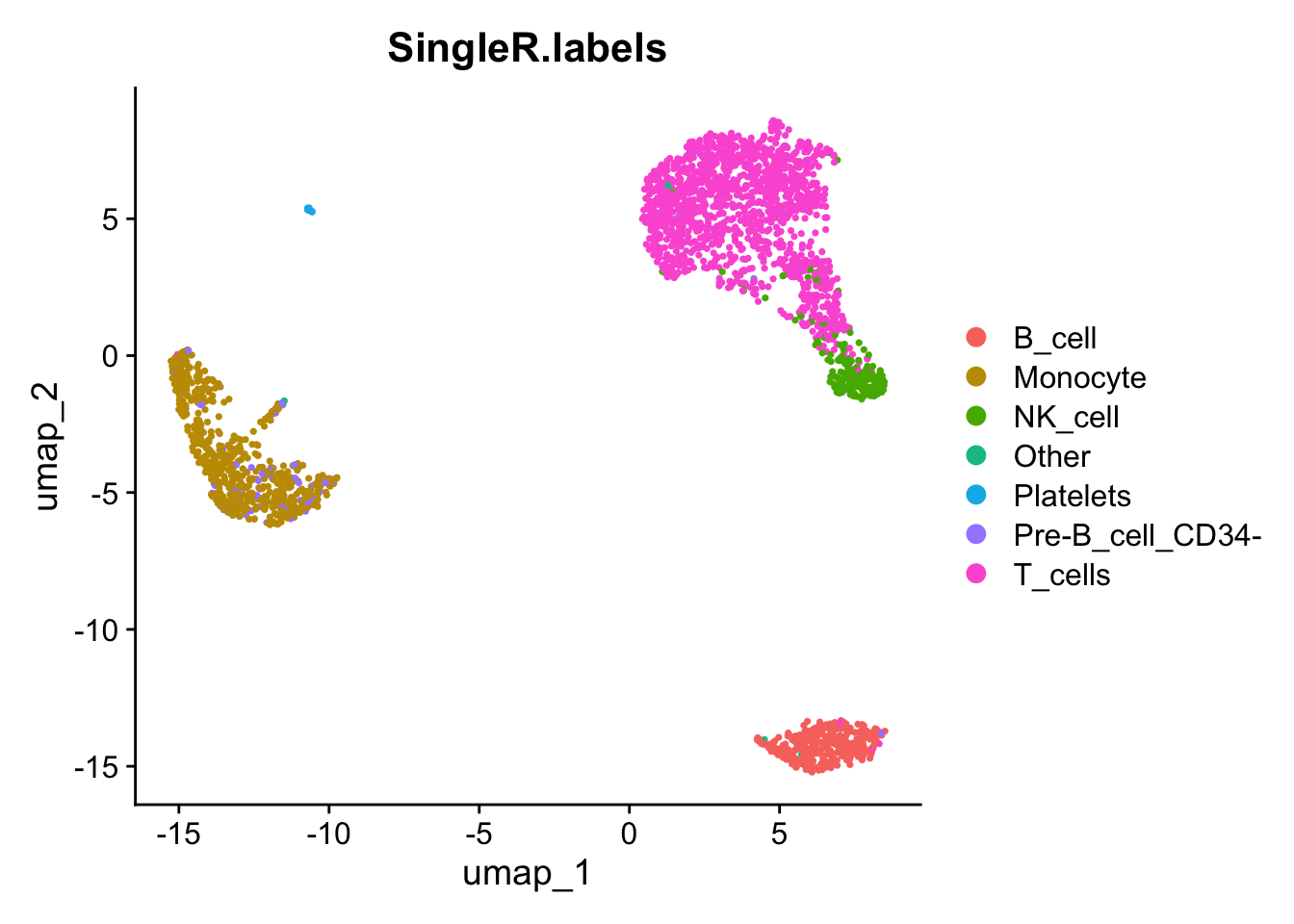
It is nice to see that even though SingleR does not use the clusters we computed earlier, the labels do seem to match those clusters reasonably well.